



La Théorie des Perturbations Fonctionnelles de la Densité (DFPT) de notre outil atomistique RESCU fournit un moyen de simuler efficacement les réponses fonctionnelles des matériaux, qui sont directement mesurées dans des expériences pour mieux comprendre un matériau. Dans nos articles précédents, nous avons montré comment RESCU-DFPT est utilisée pour calculer les réponses aux déplacements atomiques, aux champs électriques statiques et aux combinaisons des deux, c’est-à-dire effet des phonons, la constante diélectrique aux ions liés et les charges effectives de Born, respectivement. Dans cet article, nous montrons que les propriétés optiques du spectre infrarouge peuvent également être simulées.
Prédiction des propriétés optiques infrarouges avec la méthode DFPT de notre solveur RESCU
February 15th, 2026
Technologies
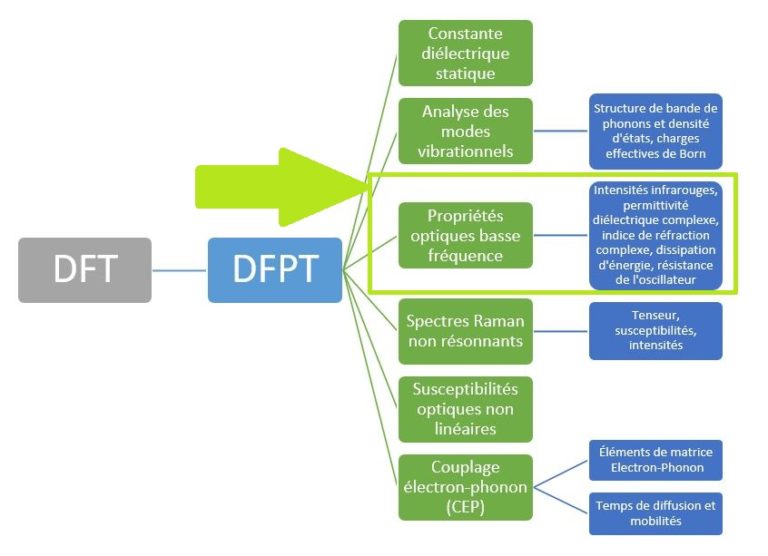
Figure 1 – Liste de nos principales fonctionnalités DFPT au sein de notre solveur RESCU.
La réponse de nombreux matériaux à un champ électromagnétique externe dans la région infrarouge est dominée par les vibrations du réseau. Les photons infrarouges ne sont pas assez énergétiques pour provoquer des transitions électroniques, mais leur énergie est suffisante pour entraîner des transitions phononiques de grande longueur d’onde. C’est pourquoi la spectroscopie infrarouge est une technique puissante pour sonder les propriétés vibrationnelles des matériaux.
Les données de spectroscopie infrarouge peuvent être corroborées et prédites par la voie théorique en simulant la physique vibrationnelle des atomes d’un réseau. Les simulations atomistiques peuvent être utilisées pour interpréter les résultats expérimentaux et nous fournissent en outre des informations qui ne peuvent pas être mesurées expérimentalement comme les fonctions d’onde. Pour illustrer cela, nous calculons et analysons les spectres optiques du nitrure d’aluminium (AlN) dans sa structure hexagonale en utilisant l’approximation de la densité locale (LDA) pour la fonctionnelle xc (échange-corrélation). Nous calculons la permittivité diélectrique complexe (dépendant de la fréquence) et dérivons l’indice de réfraction, ainsi que les intensités infrarouges le long de l’axe c du cristal : ce sont les propriétés optiques prépondérantes.
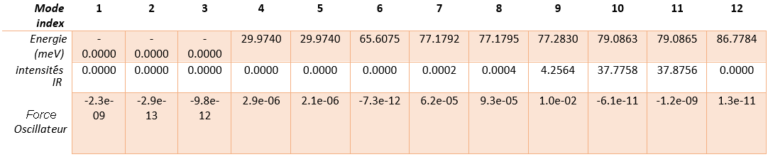
Tableau 1. Énergie calculée, intensité infrarouge et force de l'oscillateur pour tous les modes de phonons dans l'AlN hexagonal.
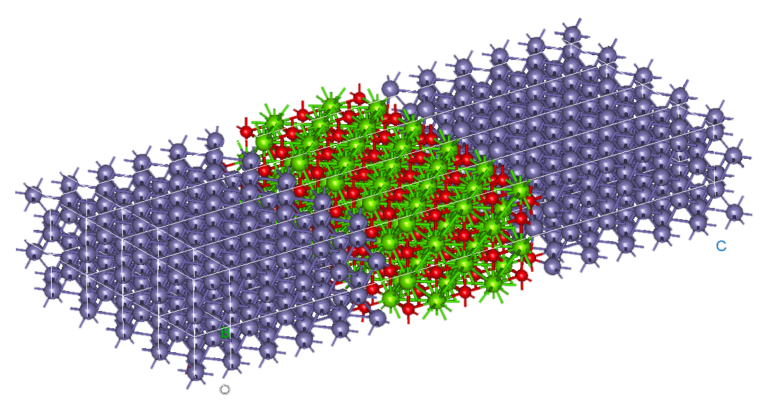
Le réseau hexagonal AlN a une cellule primitive contenant 2 atomes d’aluminium et 2 atomes d’azote. Il existe donc 12 modes vibratoires : trois acoustiques et neuf optiques. L’examen des intensités IR révèle que seuls trois modes de phonons sont IR actifs. Modes 7 & 8 (77,18 meV), modes 9 (77,28 meV) et modes 10 & 11 (79,09 meV), parmi lesquels les derniers modes ont les plus grandes intensités IR. Ces prédictions correspondent parfaitement aux intensités IR observées expérimentalement [1]. Contrairement aux expériences, les simulations donnent les déplacements atomiques de chaque mode vibrationnel, ce qui donne un meilleur aperçu. Les modes actifs IR sont représentés sur la figure 2. Par exemple, nous pouvons voir que pour les modes #7 et #8, les atomes vibrent dans le plan et dans le mode #9 ils vibrent le long de l’axe c.
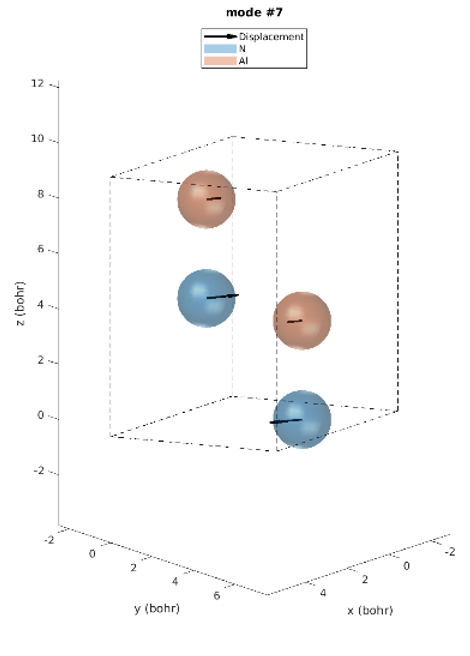
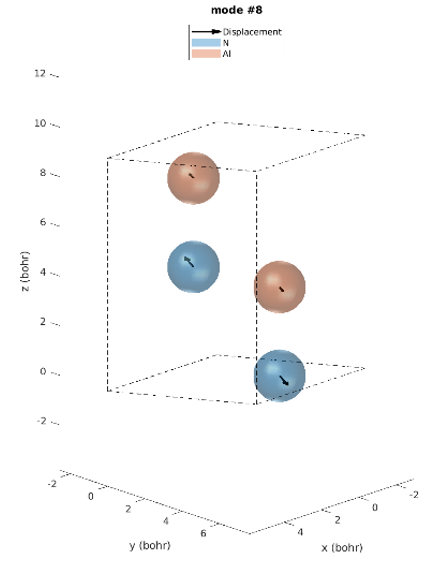
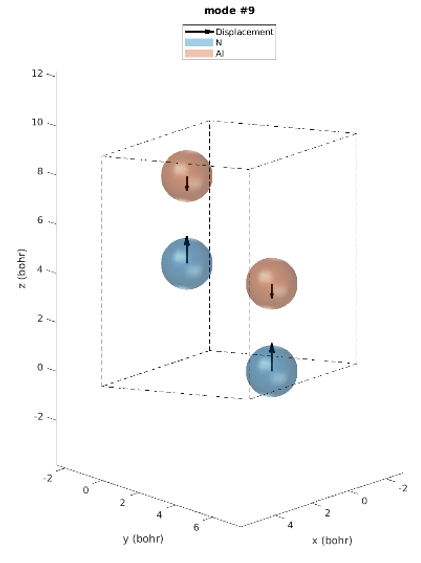
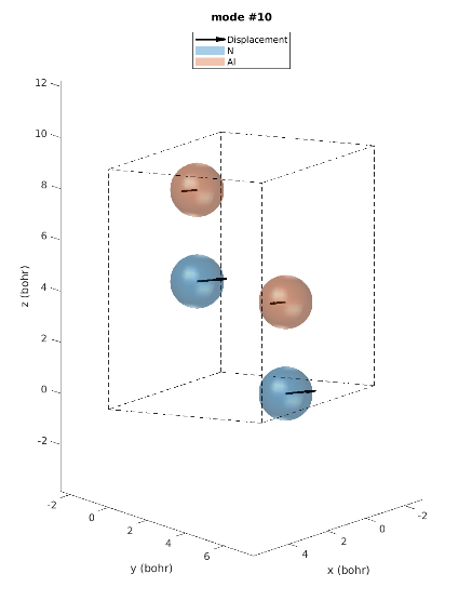
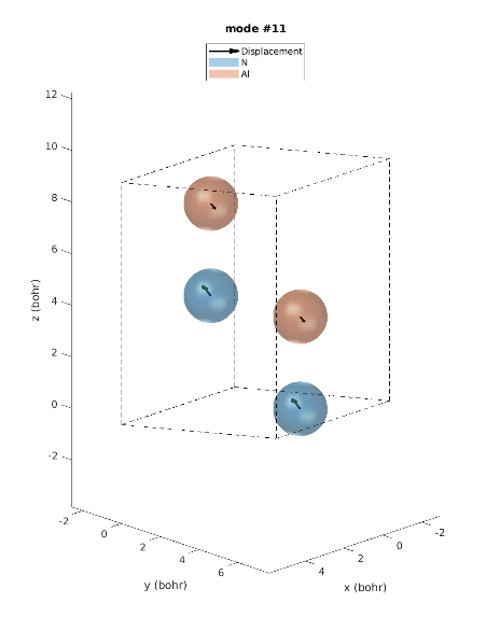
Figure 2. Modes Phononiques #7 à 11.
Nous comparons également la permittivité diélectrique et l’indice de réfraction simulés avec les données expérimentales [1] présentées dans la figure 3 ci-après. Nous prédisons 4,65 pour la constante diélectrique haute fréquence, ce qui se compare bien à la valeur expérimentale de 4,84. Les spectres expérimentaux sont extraits des spectres de réflectance en utilisant l’analyse de Kramers-Kronig. Selon ces chiffres, les simulations par DFPT capturent les principales caractéristiques des spectres optiques. De l’analyse spectrale, nous concluons que la caractéristique principale correspond à un mode phononique TO (Optique Transverse) dont la fréquence est extraite du maximum de l’indice de réfraction à 82,7 meV. Les caractéristiques spectrales dépendent directement de la force de l’oscillateur, obtenue dans les simulations, et le tableau 1 montre clairement que le mode #9 à 77,28 meV a une force d’oscillateur significativement plus grande que les autres.
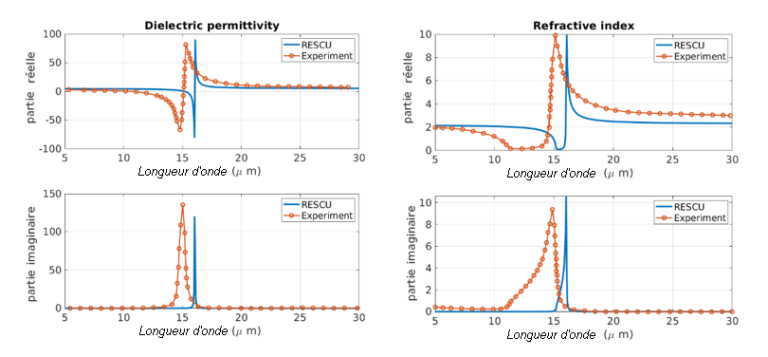
Figure 3. Permittivité diélectrique et indice de réfraction de l’AlN. Les résultats expérimentaux sont en orange et les prédictions par RESCU en bleu.
Pour résumer, plusieurs propriétés optiques d’AlN peuvent être calculées par DFPT en utilisant notre code RESCU. Ces informations peuvent être utilisées pour valider des hypothèses expérimentales, par exemple pour confirmer la pureté du matériau et sa phase dominante (AlN hexagonal dans notre exemple) ou en particulier en savoir plus sur des matériaux expérimentalement difficiles à synthétiser ou à étudier.
Dans une publication à venir ici sur LinkedIn, nous montrerons comment le module DFPT de notre outil RESCU est également utilisé pour calculer le spectre Raman et les intensités dans les régions infrarouges qui caractérisent l’interaction inélastique des photons avec les phonons de grande longueur d’onde dans les matériaux.
Nous espérons que vous avez apprécié la lecture, merci et n’hésitez pas à commenter et à nous demander comme d’habitude maintenant des informations spécifiques sur nos outils atomistiques, nous serons plus qu’heureux de vous donner des informations supplémentaires.
A bientôt !
[1] Lattice Vibration Spectra of Aluminum Nitride, A. T. Collins, E. C. Lightowlers, and P. J. Dean, Phys. Rev. 158, 833


