





Written in Python and Fortran, it comes with new Python calculators and provides extra scalability for supercomputing platforms. It is fully compatible with our new quantum transport tool NanoDCAL+.
RESCU+
RESCU+ is the newly designed and enhanced version of RESCU.
Who are the
customers using RESCU+?

Battery engineers
RESCU+ is a premium tool to do battery research. Its efficiency can be exploited to perform the complex and computationally intensive ab initio molecular dynamics (AIMD) calculations required by battery R&D faster and at lower cost.
Chemical engineers
Use RESCU+ together with the nudged elastic band (NEB) method to study the catalytic activity of interfaces or surfaces, thus accelerating the pace of experimentation and reducing R&D costs.
Materials engineers
Predict advanced and quantum material functions in silico. The capabilities of RESCU+ are particularly useful in simulating realistic materials - doped semiconductors, VdW heterostructures, alloys, heterojunctions, liquid/solid interfaces - whose unit cell contains thousands of atoms.
Academic researchers
Experimentalists and theorists use this convenient and powerful implementation of DFT to compute the ground state properties of molecules, interfaces and crystals from first principles.
Benefits
RESCU+Predict the electronic structure of virtually any material.
RESCU+ computes the properties of molecules, crystals, surfaces and large scale heterostructures from first principles using density functional theory (DFT).
Using RESCU+ is convenient and easy.
RESCU+ is operable using the popular Python language. You can conveniently build crystals, heterostructures and other systems directly in Python and import calculators to simulate and analyze atomistic systems. You can also easily create your own workflows using RESCU+ calculators.
Get the answer faster using RESCU+ powerful implementation.
RESCU+’s high-performance solvers are carefully optimized and parallelized to yield the answer faster and allow simulating larger more realistic systems (up to 100,000+ atoms).
Efficient ion dynamics
The RESCU+ python API makes it easy to interface with other libraries such as ASE and LAMMPS, which can use RESCU+ to quickly do away with large scale relaxation, AIMD and NEB calculations.
Download the RESCU+ leaflet to get a summary of the software features!

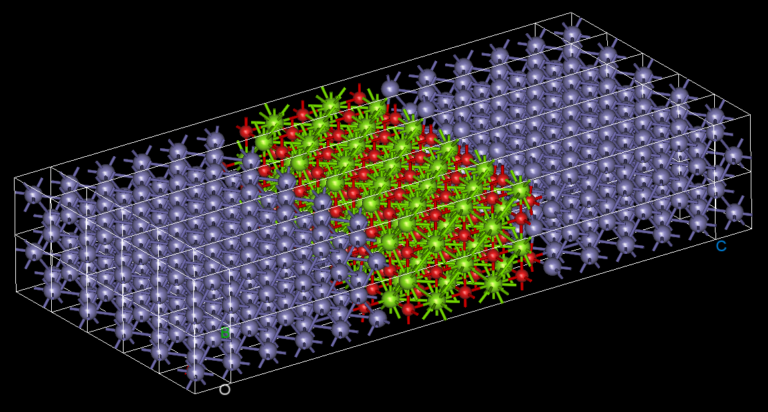
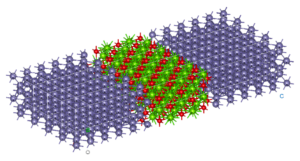
Twisted bilayer graphene’s small rotation angle leads to long range order resulting in a primitive cell with more than 2,000 atoms.
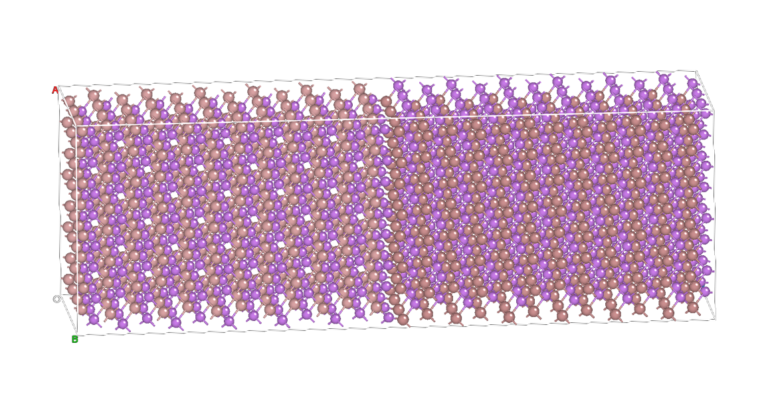
Realistic battery materials include many elements resulting in complex chemical and structural interactions.
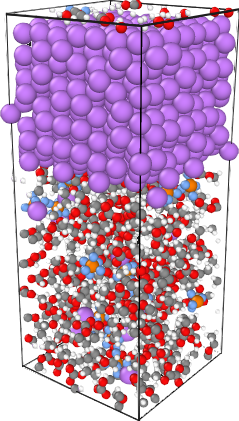
A Ti-H complex processing on an MgO surface might explain the observed large g-factor anisotropy.
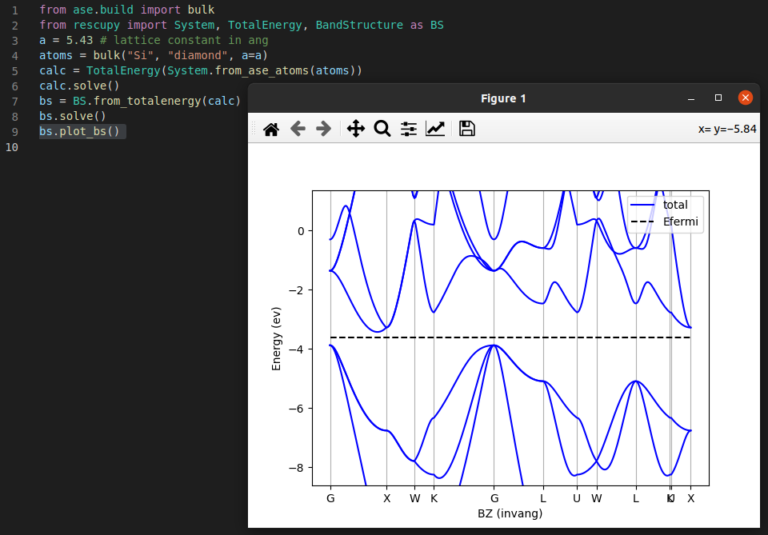
Create materials and complex workflows, and perform atomistic simulations with simple Python scripts.
Features
RESCU+Description
RESCU+ – Real space Electronic Structure CalcUlator – is a powerful density functional theory (DFT) solver, entirely written in Fortran at the core and with a Python interface. It can predict the electronic structure and derived properties of bulk materials, material surfaces and molecules. RESCU+ calculates the ground-state density using a basis of numerical atomic orbitals. Written with the objective of solving systems comprising tens of thousands of atoms, RESCU+ is carefully parallelized and exploits libraries such as FFTW, MPI, ScaLAPACK and others. It includes many state-of-the-art analysis tools such as density of states (DOS), projected density of states (PDOS), local density of states (LDOS, PLDOS) and band structure tools. It is fully compatible and integrated with our new quantum transport tool NanoDCAL+.
Fast & parallel solver
RESCU+ is carefully optimized to get you the answer faster. The code is parallelized using MPI and scales to 1,000’s of cores.

Python integration
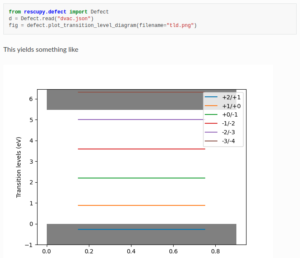
RESCU+ has a friendly Python interface, allowing one to quickly and easily build workflows and visualize data. Choose from our pool of calculators to compute ground state densities, band structures, equations of states, transition level diagrams of defects and more.
Ion dynamics
Quickly find equilibrium atomic positions and cell shapes of large structures, perform molecular dynamics and nudged elastic band calculations using RESCU+ as a powerful first principles force and stress calculator. RESCU+ harbors a hybrid ab initio/machine learning (ML) molecular dynamic engine which can accelerate AIMD workloads by more than an order of magnitude.
Ground state properties
Predict ground state properties like total energy, atomic forces, stress tensor.

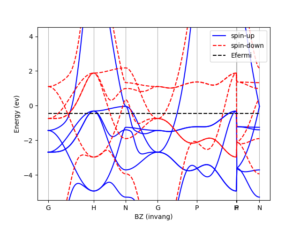
Spin
Include the physics of electronic spin and spin-orbit coupling via a state-of-the-art spin-DFT implementation (collinear and non-collinear formalisms).
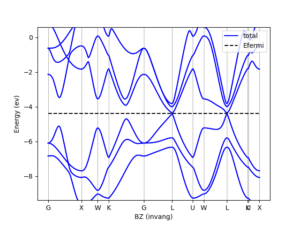
Atomic orbitals and pseudopotentials
Benefit from our accurate, efficient and complete database of atomic orbitals and pseudopotentials.
What's new ?
We have just released RESCU+, please contact us to discuss your needs and benefit from a special license offer to celebrate!
Current RESCU+ version is 2023B.
RESCU+ user documentation
To access to RESCU+ user documentation, click the link below: installation and user manuals, tutorials, theory and technical information pages are all there for your support and to get you up to speed as quickly as possible.