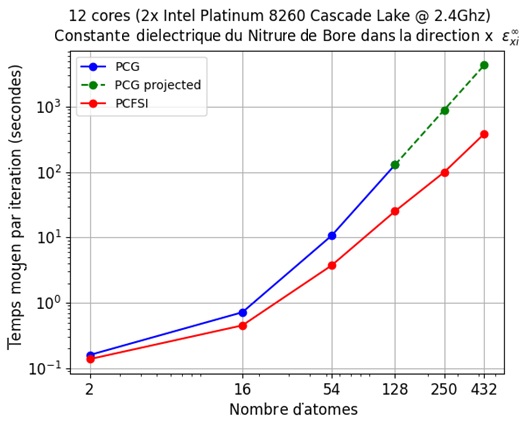
Figure 3 – Les gains de temps de calcul obtenus par notre méthode PCFSI vs taille des supercellules qui augmente.
Ce résultat obtenu pour le BN peut être généralisé à d’autres types de matériaux : avec l’augmentation de la taille des cellules, notre méthode devient prédominante par rapport à toute autre méthode comparable.
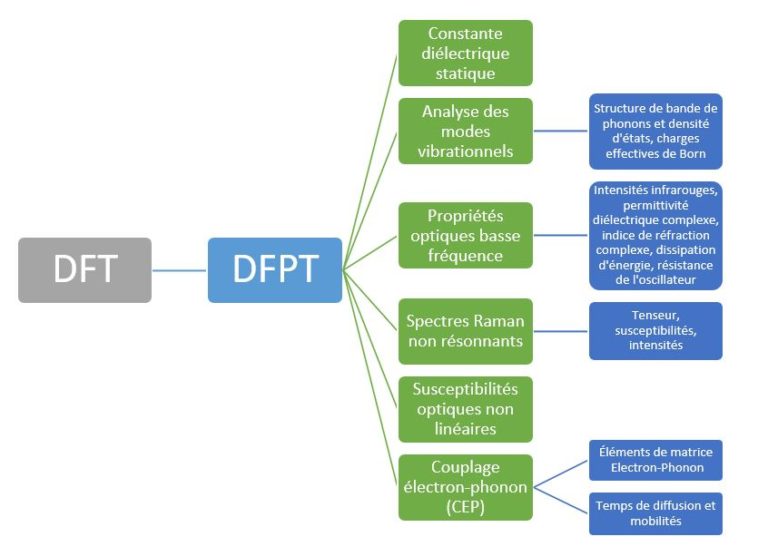
Nous voyons que la méthode PCFSI surpasse déjà le PCG en utilisant 12 cœurs pour une cellule de taille unitaire et l’écart se creuse rapidement à mesure que la taille du système augmente, de sorte qu’il est environ 8 fois plus rapide pour les supercellules à 128 atomes. Parce que la PCFSI a des besoins inférieurs en mémoire, elle peut être utilisée pour simuler des supercellules jusqu’à 432 atomes tandis que le solveur PGC ne pouvait simuler que des systèmes avec 128 atomes maximum dans le test d’étalonnage ci-dessus. Plus de détails sur l’algorithme PCFSI (matrice dynamique, structure de bande et densité d’états phononiques, spectre Raman, etc.) seront donnés dans nos prochaines publications ici sur LinkedIn : alors pour ne pas les manquer, nous vous invitons à nous suivre pour être informés de notre actualité. Nous montrerons que celui-ci peut être adapté pour calculer toutes les réponses fonctionnelles typiquement obtenues par DFPT.
Nous espérons que vous avez apprécié la lecture de cet article, merci et n’hésitez pas à commenter et à nous demander des informations spécifiques sur nos outils atomistiques et méthodes mises en œuvre, nous serons plus qu’heureux de vous donner des informations supplémentaires et vous faire tester nos outils.
Plus de contenu sur la science de la simulation à venir très bientôt !
[1] Zhou, Y., Saad, Y., Tiago, M., & Chelikowsky, J. (2006). Parallel self-consistent-field calculations via Chebyshev-filtered subspace acceleration. Physical Review E, 74(6), 066704. https://doi.org/10.1103/PhysRevE.74.066704
[2] Michaud-Rioux, V., & Guo, H. (2017). RESCU: extending the realm of Kohn-Sham density functional theory. https://escholarship.mcgill.ca/concern/theses/dn39x387d?locale=en
[3] Bohloul, S. (2017). First-Principles Quantum Transport and Linear Response Modeling of Nano-devices and Materials. https://escholarship.mcgill.ca/concern/theses/8910jx167?locale=en
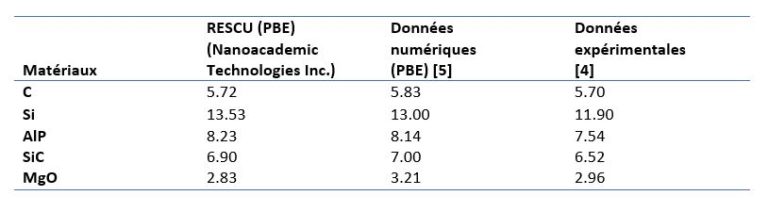
[4] Gajdoš, M., Hummer, K., Kresse, G., Furthmüller, J., & Bechstedt, F. (2006). Linear optical properties in the projector-augmented wave methodology. Physical Review B – Condensed Matter and Materials Physics, 73(4), 045112. https://doi.org/10.1103/PhysRevB.73.045112
[5] A. Jain*, S.P. Ong*, G. Hautier, W. Chen, W.D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder, K.A. Persson (*=equal contributions), The Materials Project: A materials genome approach to accelerating materials innovation, APL Materials, 2013, 1(1), 011002.