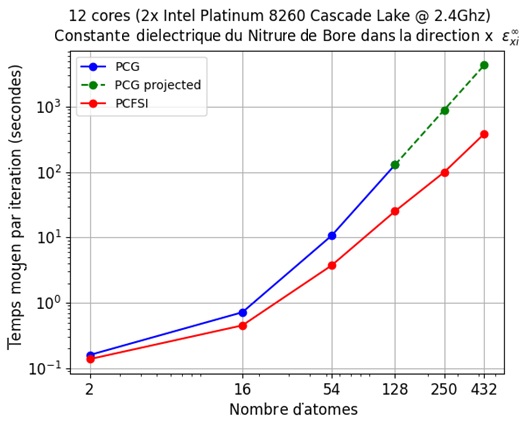
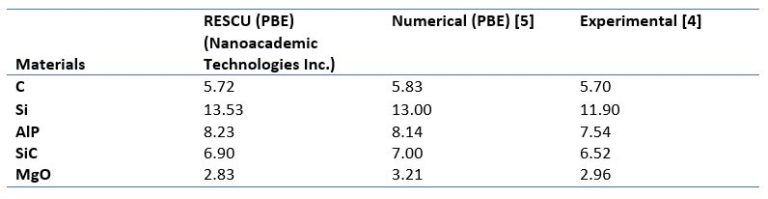
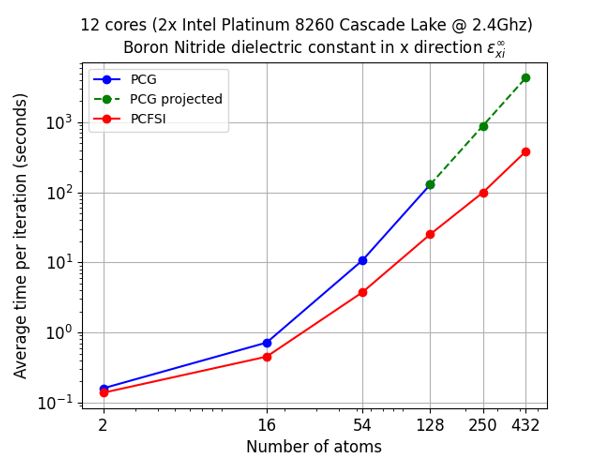
Figure 3 – The computational time gains of our PCFSI method while supercell sizes increase.
This result obtained for BN can be generalized to other types of materials: with increasing cell sizes our method becomes prevalent Vs any other methods in those case studies.
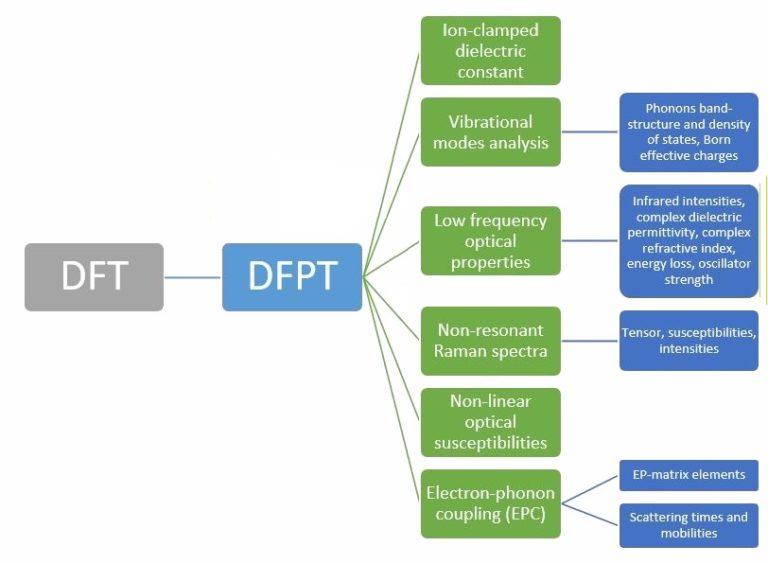
We see that PCFSI already outperforms PCG using 12 cores for all unit cell sizes and the gap is growing rapidly as system size increases, such that it is around 8 times faster for supercell with 128 atoms. Because PCFSI has lower memory requirements, it can be used to simulate supercells up to 432 atoms while the PGC solver could only simulate systems with 128 atoms in the above benchmark. More details on the PCFSI algorithm (dynamical matrix, phonon band-structure and density of states, Raman spectrum, and more) will be explained in forthcoming publications here on LinkedIn, so do not miss them! We will show it can be adapted to compute all response functions typically obtained in DFPT.
We hope you enjoyed the read, thank you and feel free to comment and ask us specific information about our atomistic tools, we will be more than happy to give additional insights.
More simulation science content to come very soon! Please stay tuned.
[1] Zhou, Y., Saad, Y., Tiago, M., & Chelikowsky, J. (2006). Parallel self-consistent-field calculations via Chebyshev-filtered subspace acceleration. Physical Review E, 74(6), 066704. https://doi.org/10.1103/PhysRevE.74.066704
[2] Michaud-Rioux, V., & Guo, H. (2017). RESCU: extending the realm of Kohn-Sham density functional theory. https://escholarship.mcgill.ca/concern/theses/dn39x387d?locale=en
[3] Bohloul, S. (2017). First-Principles Quantum Transport and Linear Response Modeling of Nano-devices and Materials. https://escholarship.mcgill.ca/concern/theses/8910jx167?locale=en
[4] Gajdoš, M., Hummer, K., Kresse, G., Furthmüller, J., & Bechstedt, F. (2006). Linear optical properties in the projector-augmented wave methodology. Physical Review B – Condensed Matter and Materials Physics, 73(4), 045112. https://doi.org/10.1103/PhysRevB.73.045112
[5] A. Jain*, S.P. Ong*, G. Hautier, W. Chen, W.D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder, K.A. Persson (*=equal contributions), The Materials Project: A materials genome approach to accelerating materials innovation, APL Materials, 2013, 1(1), 011002.