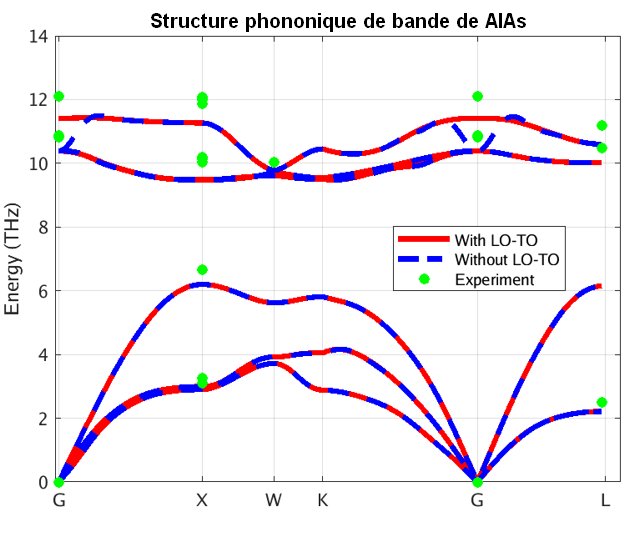
Figure 3 – Structure de bandes phononique de l’Arseniure d’Aluminium calculée avec notre méthode DFPT
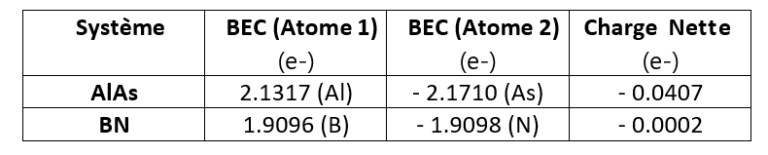
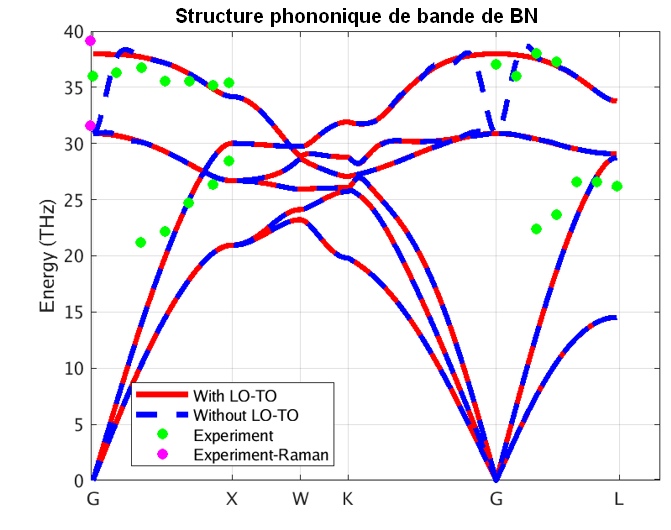
Sur ces figures, les lignes bleues et rouges représentent les énergies de phonons calculées respectivement sans et avec effet des BEC. La division des énergies des phonons pour les modes optiques transversaux et longitudinaux (TO et LO) est aussi clairement visible en comparant les structures des bandes bleue et rouge. Le dédoublement LO-TO peut également être observé dans les données expérimentales (illustrées en vert et violet) où l’effet lève la dégénérescence des modes LO et TO (BN [2] et AlAs [3]). Comme indiqué ci-dessus, cela est dû au champ électrique induit des BEC qui exercent une force supplémentaire sur les atomes en mode LO. Les énergies des modes LO dans AlAs et BN sont augmentées d’environ 1 et 7 THz respectivement, ce qui constitue une correction significative.
Notre simulation PBE capture les informations essentielles concernant les spectres de phonons pour ces systèmes, y compris la division LO-TO. Il s’avère que le spectre de phonons de la LDA (Local Density Approximation) d’AlAs et de BN (non illustré ici) se compare mieux aux données expérimentales rapportées. Notons cependant que les mesures expérimentales sont rares et dispersées autour des prédictions théoriques. De plus, les simulations théoriques prédisent correctement le comportement linéaire des phonons acoustiques (raies Γ-X et Γ-L proches de Γ), contrairement aux mesures expérimentales. Ceci témoigne de la difficulté de mesurer les spectres de phonons et motive le besoin d’un codes de principes premiers comme RESCU qui soit précis et efficaces pour parvenir à cela.
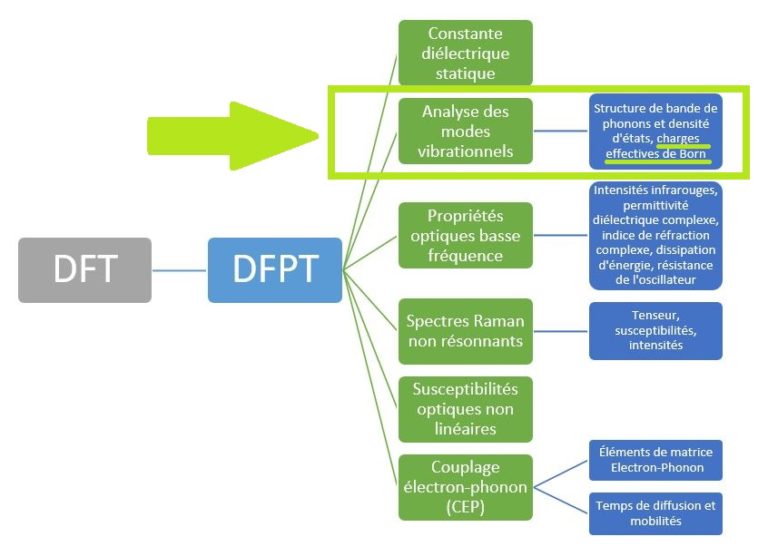
Dans une publication à venir ici sur LinkedIn, nous montrerons comment le module DFPT utilise les ingrédients susmentionnés pour calculer les propriétés optiques telles que les intensités infrarouges, la permittivité diélectrique dépendante de la fréquence et l’indice de réfraction.
Nous espérons que vous avez apprécié cette brève lecture, merci et n’hésitez pas à commenter et à nous demander des informations spécifiques sur nos outils atomistiques, nous serons plus qu’heureux de vous donner des informations supplémentaires comme à l’accoutumée.
Plus de contenu sur la science de la simulation est définitivement au coin de la rue, avec des nouvelles prochainement sur nos prochains outils et fonctionnalités, alors suivez-nous et connectons-nous pour ne rien manquer des prochains articles !
[1] Xavier Gonze and Changyol Lee (1997). Dynamical matrices, Born effective charges, dielectric permittivity tensors, and interatomic force constants from density-functional perturbation theory: Phys. Rev. B 55, 10355 – Published 15 April 1997
[2] Hage, F. S., Nicholls, R. J., Yates, J. R., McCulloch, D. G., Lovejoy, T. C., Dellby, N., Krivanek, O. L., Refson, K., & Ramasse, Q. M. (2018). Nanoscale momentum-resolved vibrational spectroscopy. Science Advances, 4(6), eaar7495. https://doi.org/10.1126/SCIADV.AAR7495
[3] Azuhata, T., Sota, T., & Suzuki, K. (1995). Second-order Raman spectra and lattice dynamics in AlAs. Journal of Physics: Condensed Matter, 7(9), 1949. https://doi.org/10.1088/0953-8984/7/9/018