* SCF is the Self-Consistence Field iterative method.
As illustrated, typical KS-DFT codes take in the range of hours to solve 1k-atom systems, while we require on average 10 minutes only with RESCU. We gain another order of magnitude when the ML engine kicks in.
What does our ML engine do?
- Predicts energies and atomic forces based upon multi-fidelity results (low-fidelity to high-fidelity fast mapping);
- Its model predictions are designed to produce chemical-accurate results;
- Its models are adaptively upgraded on the fly during the AIMD calculation.
Note: processing was made in collaboration with McGill University on the Niagara cluster and we estimate that the ML part represented 90% of the effort and the 10% remaining was pure DFT.
For large systems (> 100 atoms but even more true for 1k atoms and beyond), our codes have the ability to quickly converge thus will save you a huge amount of time allowing you to simulate more, try more configurations and more complex ones, than with your current tool and limitations.
Note that a paper is about to be published about the full experiment with complete details and physical/computing information plus more insights about how our ML works and our next topics of study. Please follow us to get the latest news on our tools and results we got from our own studies or collaborations.
You may wonder why we performed such a study:
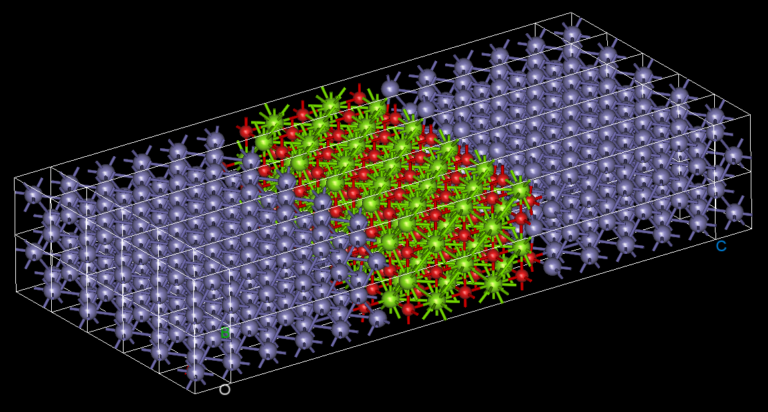
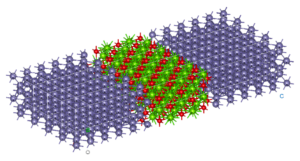
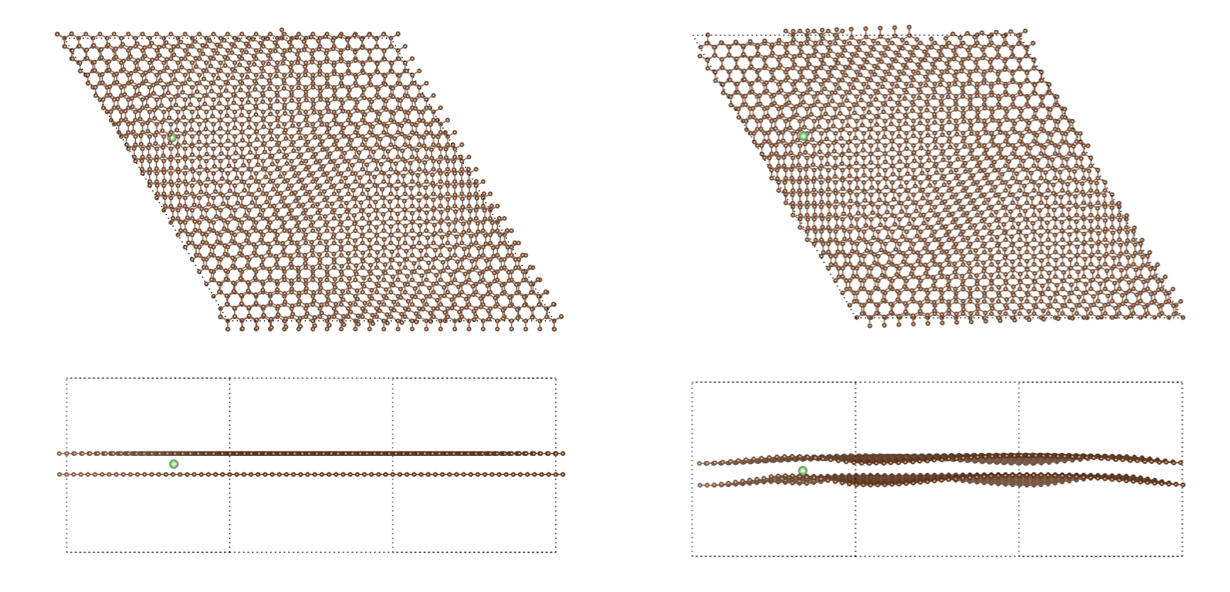
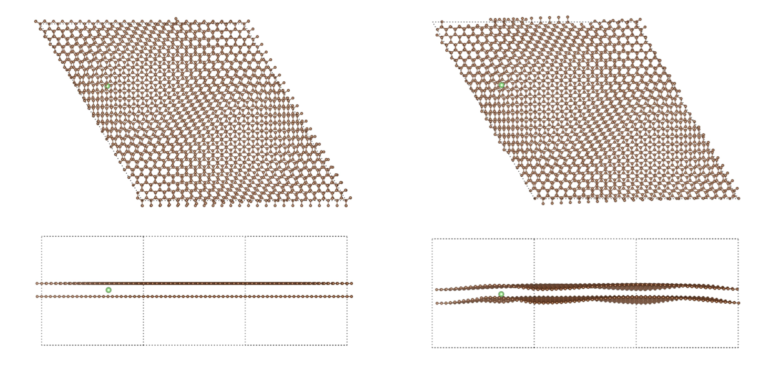
The reasons why we use twisted bilayer graphene with lithium intercalation are:
1) the twisted bilayer graphene is hot spot research field recently and relaxation of this kind of systems with pure DFT is a very difficult problem.
2) Lithium intercalation relates to Li-ion battery technologies and it is a good example for the battery industry researchers and similar such R&D.
Also, we will publish regularly articles about the features of our advanced atomistic tools (especially how they compare against other academic or commercial codes regarding material parameters accuracy and convergence time of small to very large system models), upcoming software releases, updates and miscellaneous news about Nanoacademic Technologies.