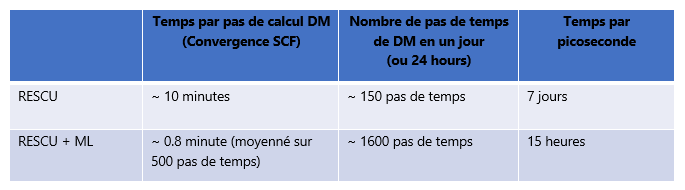
* SCF réfère à la méthode itérative du champ auto-cohérent.
Comme illustré, les codes KS-DFT typiques prennent de l’ordre de quelques heures pour résoudre des systèmes à 1 000 atomes, alors que nous n’avons besoin en moyenne que de 10 minutes avec RESCU (voire moins dans ce cas). Nous gagnons un autre ordre de grandeur lorsque le moteur d’auto-apprentissage rentre en action.
À quoi sert ce moteur spécifique ?
- Il prédit les énergies et les forces atomiques en fonction des résultats multi-précision (cartographie rapide de basse fidélité à haute fidélité) ;
- Ses prédictions de modèles sont conçues pour produire des résultats chimiquement précis;
- Ses modèles sont mis à jour de manière adaptative à la volée pendant le calcul de l’AIMD.
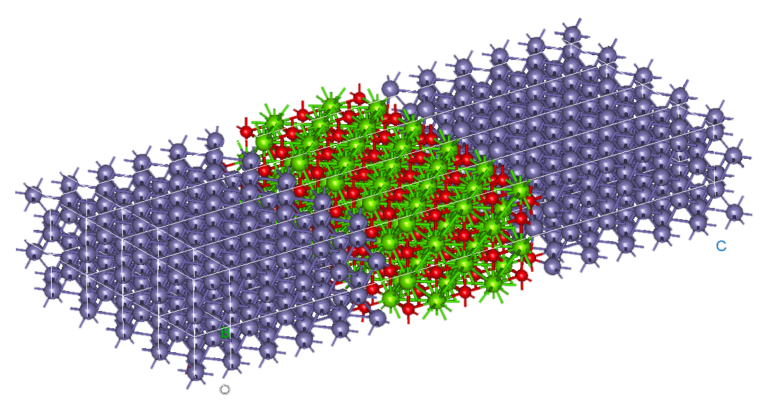
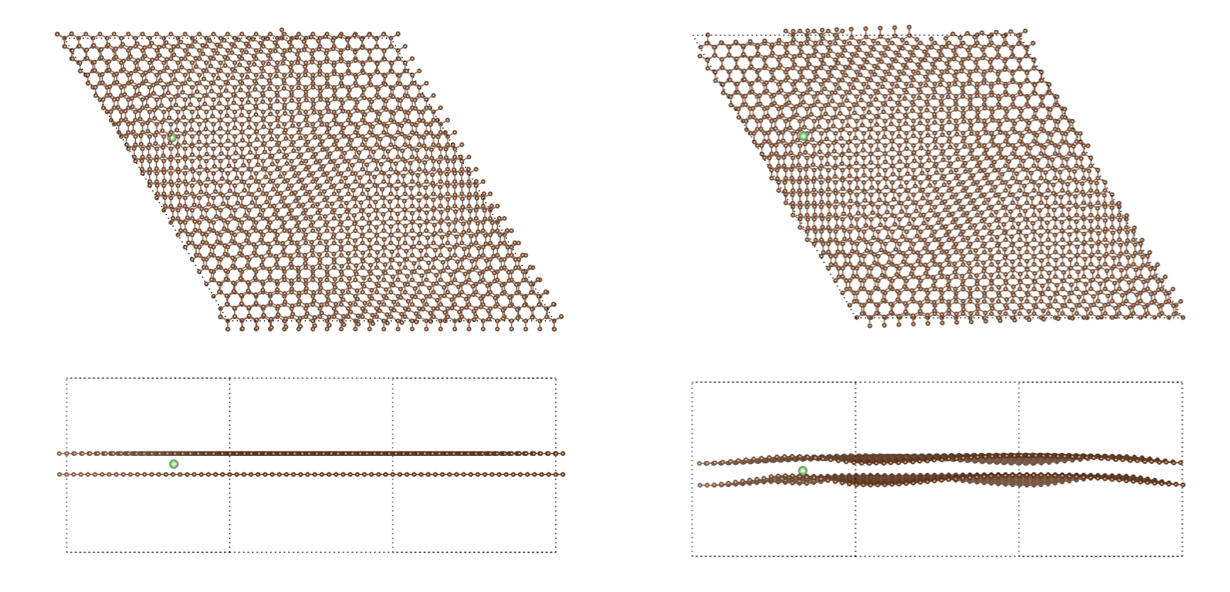
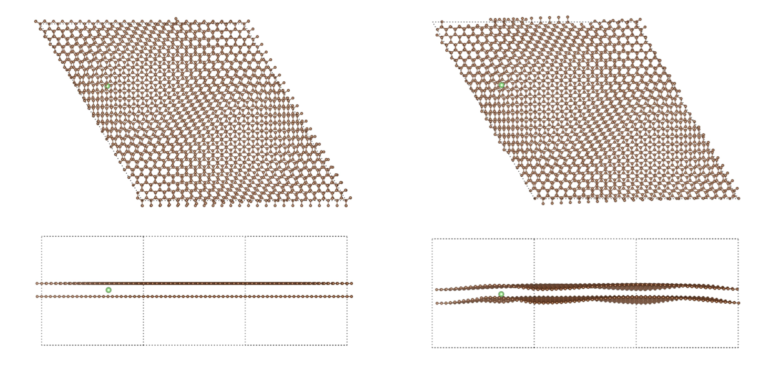
Note : le calcul a été fait en collaboration avec l’Université McGill sur la grappe Niagara et nous estimons que la partie ML représentait 90% de l’effort et les 10% restants étaient de la pure DFT.
Pour les grands systèmes (> 100 atomes mais encore plus vrai pour 1,000 atomes et au-delà), nos codes ont la capacité de converger rapidement et ainsi vous feront gagner un temps considérable vous permettant de simuler plus, d’essayer plus de configurations et possiblement plus complexes, comparativement aux limites avec lesquelles vous devez composer actuellement.
Un article est sur le point d’être publié sur cette étude avec les détails complets et des informations physiques ainsi qu’informatiques, démontrant le bon fonctionnement de notre moteur de ML et un aperçu de nos prochains sujets d’étude.
Suivez-nous pour obtenir les dernières nouvelles sur nos outils de simulations atomistiques avancées et d’autres résultats que nous avons obtenus de nos propres études ou à travers nos collaborations.