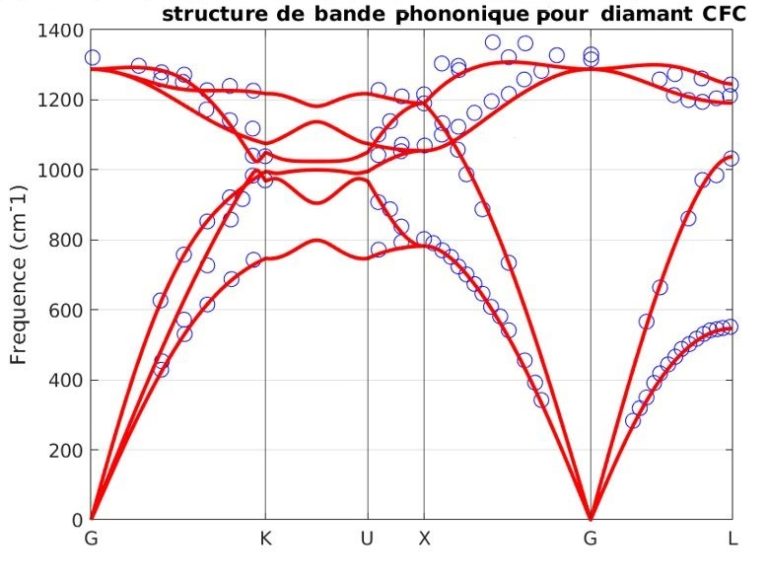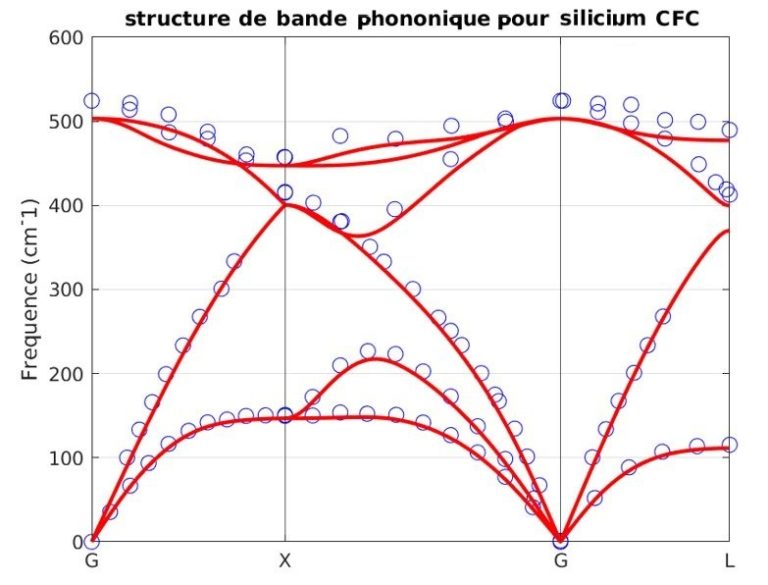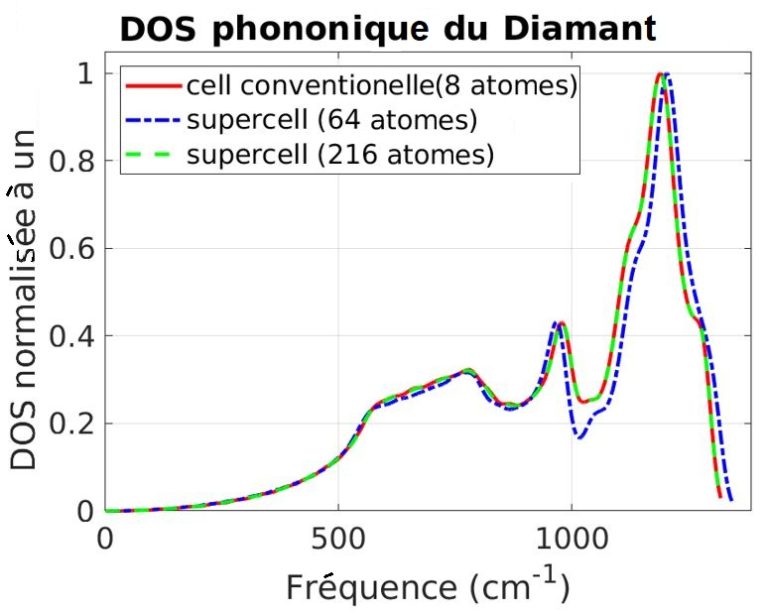




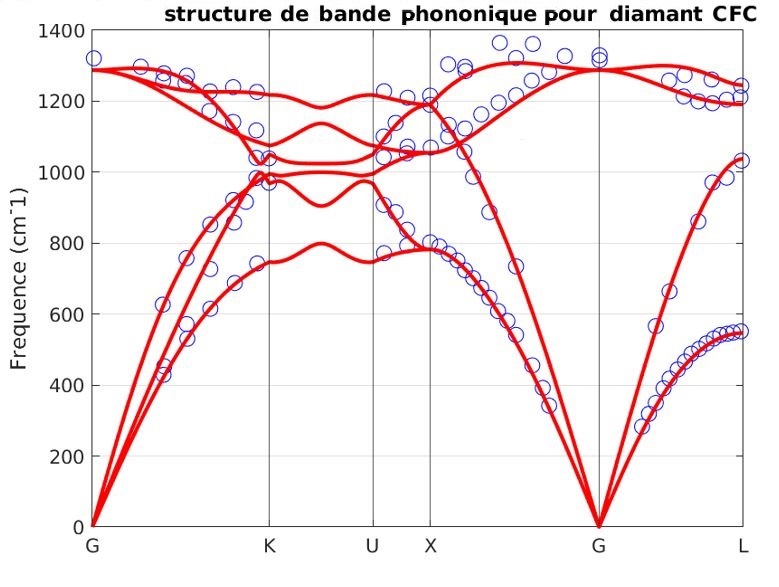
Notre outil atomistique RESCU est un puissant solveur KS-DFT (Kohn-Sham) qui résout l’équation KS sur des grilles spatiales réelles en utilisant la méthode d’itération sous-spatiale filtrée de Chebyshev (CFSI). Cette méthode CFSI évolue favorablementt avec le nombre d’atomes et les ressources de calcul disponibles, ce qui permet à RESCU de simuler de grands systèmes qui sont hors de portée des solveurs conventionnels.
Dans notre précédent article posté fin Juin, nous avions présenté notre boîte à outils de la théorie de la fonctionnelle de la densité perturbée (DFPT) et avions aussi introduit la méthode dite CFSI perturbée (PCFSI) comme notre solution unique pour les simulations DFPT à grande échelle. Nous avons démontré comment cela permet de calculer la constante diélectrique ionique (ou statique) aussi précisément que les techniques DFPT de pointe, tout en étant plus efficace et en nous permettant de simuler des systèmes jusqu’à plusieurs centaines d’atomes. Pour rappel ou pour ceux qui ont raté nos publications précédentes, l’ensemble actuel des fonctionnalités de la boîte à outils DFPT est résumé dans le schéma suivant (Figure 1) :