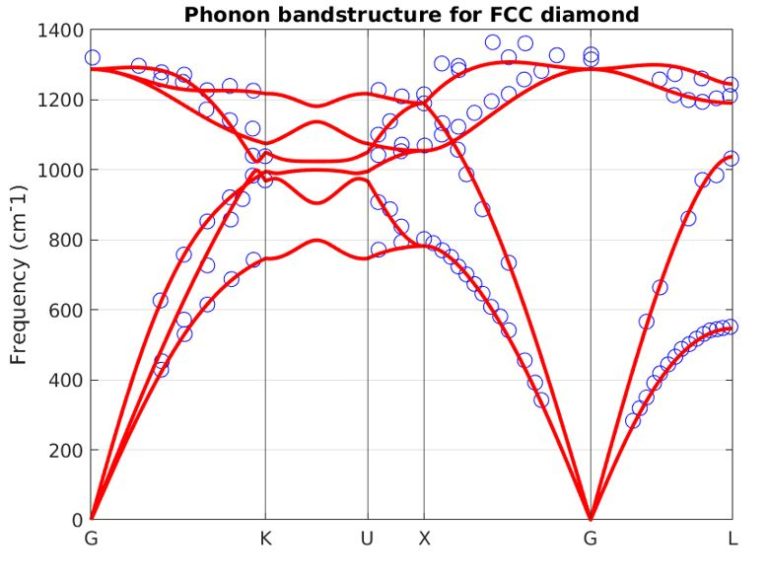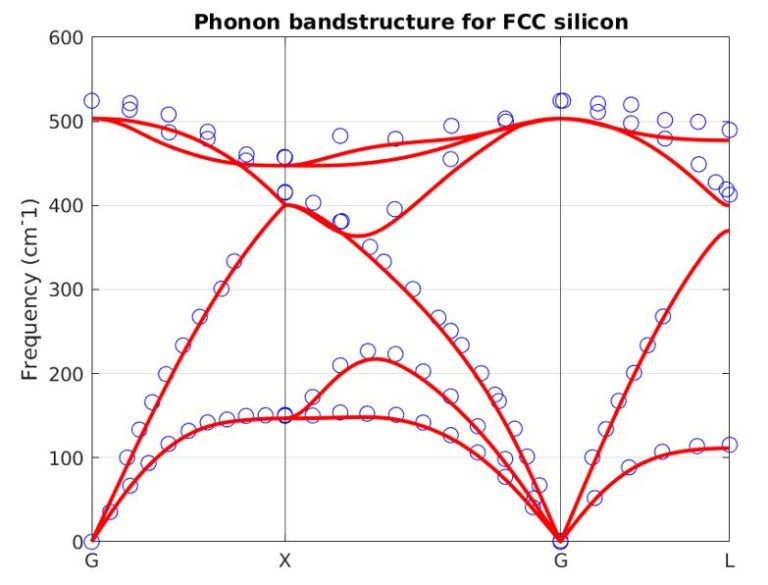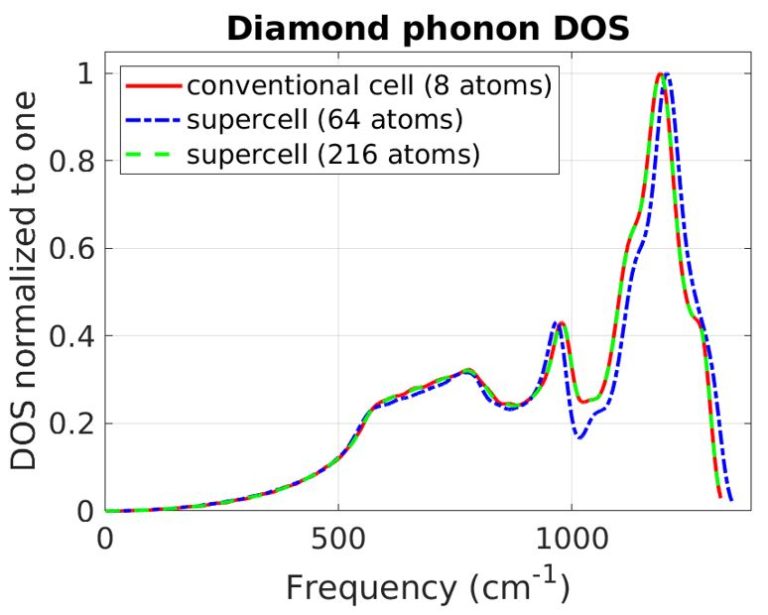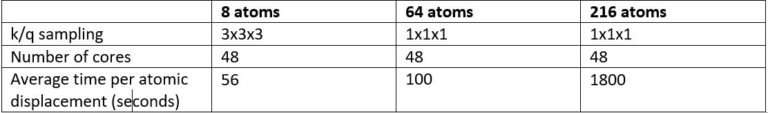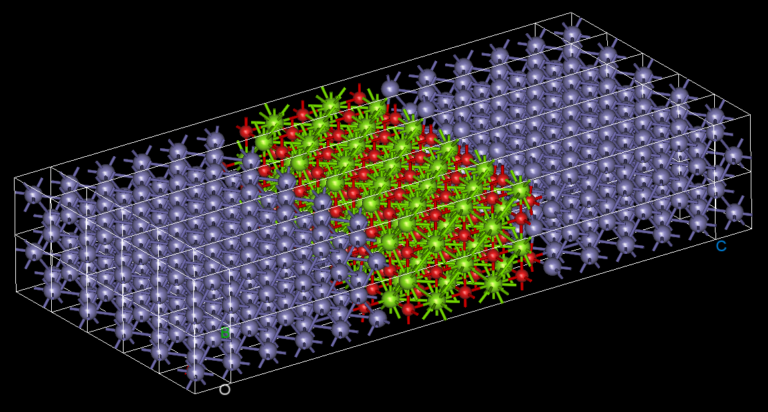





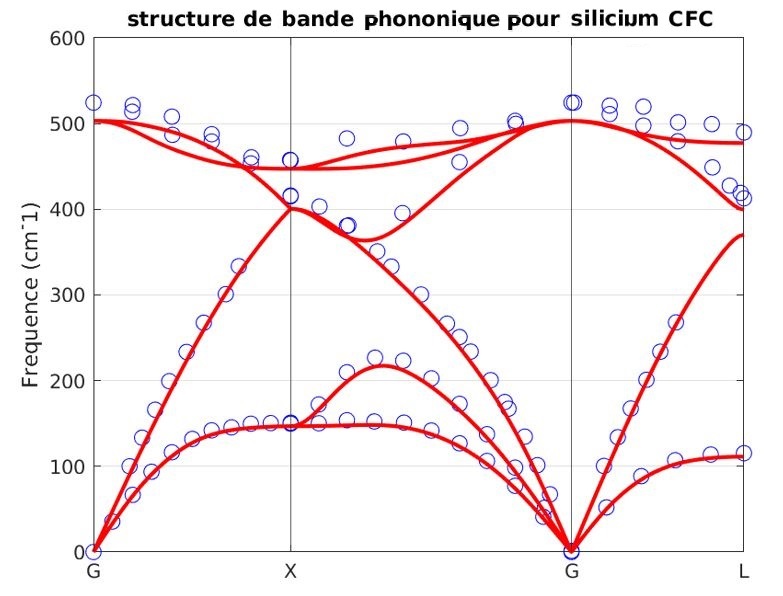
Our atomistic tool RESCU is a powerful KS-DFT solver (Kohn-Sham), which solves the KS equation on real space grids by using the Chebyshev filtered subspace iteration (CFSI) method. CFSI scales efficiently with the number of atoms and computational resources which empowers RESCU to simulate large systems which are beyond the reach of conventional solvers.
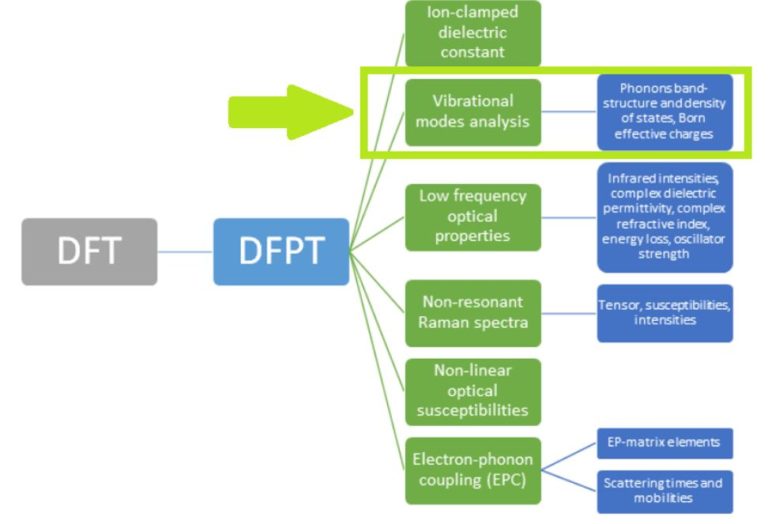
In our previous LinkedIn post, we introduced our density functional perturbation theory (DFPT) toolbox and the so-called perturbed CFSI (PCFSI) method, as our unique solution for large scale DFPT simulations. We demonstrated how it can compute the ion-clamped (or static) dielectric constant as precisely as state-of-the-art DFPT techniques, while being more efficient and allowing us to simulate systems up to several hundreds of atoms. As a reminder or for those who have missed our previous postings, the current set of features of DFPT toolbox is summarized in the following diagram (Figure 1):