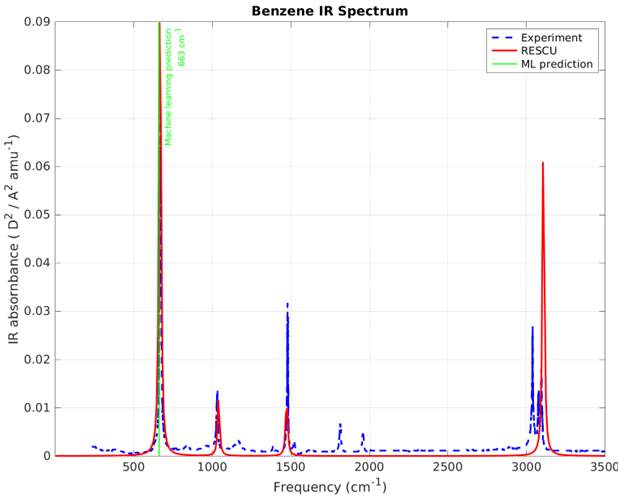
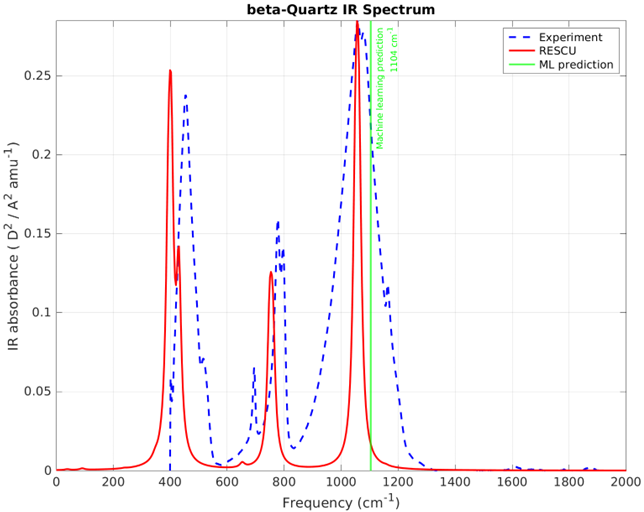
Dans cet article, nous avons montré comment RESCU peut être utilisé comme outil précis pour la simulation atomistique des spectres IR pour différents types de matériaux assez représentatifs, que ce soit pour une meilleure compréhension des données expérimentales disponibles ou comme seule solution pour mieux comprendre dans les cas où ces données ne sont pas disponibles. En effet, les expérimentateurs et les théoriciens trouvent de grands avantages à utiliser nos solutions atomistiques.
Nanoacademic Technologies propose des licences commerciales RESCU pour les utilisateurs individuels et les groupes de recherche avec différentes possibilités et options d’abonnement. Pour plus d’informations, veuillez consulter notre site Web et notre portail de documentation où vous pouvez également trouver plus d’informations sur les fonctionnalités de RESCU. Pour un essai gratuit de 30 jours, veuillez-vous inscrire sur notre portail utilisateur où vous pourrez le télécharger. La méthode ML présentée ci-dessus est disponible pour notre communauté de chercheurs en consultation ou en interfaçage avec la licence RESCU pour corroborer leurs résultats expérimentaux et ceux simulés dans le cadre de leurs projets de R&D. Il s’agit d’une manière vraiment innovante d’effectuer de l’ingénierie des matériaux en économisant beaucoup de temps et de ressources dans le processus de conception.
Veuillez nous contacter pour plus d’informations. À bientôt !
Bibliographie:
[1] Baroni, Stefano, Stefano De Gironcoli, Andrea Dal Corso, and Paolo Giannozzi. « Phonons and related crystal properties from density-functional perturbation theory. » Reviews of modern Physics 73, no. 2 (2001): 515. https://doi.org/10.1103/RevModPhys.73.515
[2] Xavier Gonze and Changyol Lee (1997). Dynamical matrices, Born effective charges, dielectric permittivity tensors, and interatomic force constants from density-functional perturbation theory: Phys. Rev. B 55, 10355 – Published 15 April 1997 https://doi.org/10.1103/PhysRevB.55.10355
[3] Bohloul, S. (2017). First-Principles Quantum Transport and Linear Response Modeling of Nano-devices and Materials. https://escholarship.mcgill.ca/concern/theses/8910jx167?locale=en
[4] Kang P, Liu Z, Abou-Rachid H, et al. Machine-learning assisted screening of energetic materials[J]. The Journal of Physical Chemistry A, 2020, 124(26): 5341-5351.
[5] Liu Z L, Kang P, Zhu Y, et al. Material informatics for layered high-TC superconductors[J]. APL Materials, 2020, 8(6): 061104.
[6] Nonlinear optical susceptibilities, Raman efficiencies, and electro-optic tensors from first-principles density functional perturbation theory, M. Veithen, X. Gonze, Ph. Ghosez, Phys. Rev. B 71, 125107 (2005) https://doi.org/10.1103/PhysRevB.71.125107
[7] Spectral Database for Organic Compounds SDBS https://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi
[8] RRUFF Project https://rruff.info/