



Credits:
By Dr. Saeed Bohloul, Dr. Vincent Michaud-Rioux, Dr. Raphael Prentki, and Mr. Jeremy F. Garaffa, MSc
By Dr. Saeed Bohloul, Dr. Vincent Michaud-Rioux, Dr. Raphael Prentki, and Mr. Jeremy F. Garaffa, MSc

Infrared (IR) spectroscopy is an invaluable tool for characterizing chemical constituents of materials. This method can extract information regarding vibrations of atoms in a non-destructive way, which provides a structural fingerprint that can be used to identify materials. The main characteristic of this fingerprint is the appearance of peaks in the spectrum at certain frequencies which are unique to corresponding material. In fact, IR spectrum determines if materials hold certain properties, and hence it is used as a guideline in search of new materials with specific characteristics of interest such as molecular properties, structures, electronic densities, etc.
Accurate atomistic simulations are complementary in interpreting experimentally obtained spectra and, in some cases, are the only source of information when experimental data is not available or difficult to obtain. Despite their exceptional predictive power, atomistic simulations are not always practical tools to search the infinite material phase space for screening materials (for example based on their IR properties), due to their non-negligible computational cost. In fact, approaches based on machine learning (ML) and materials informatics (MI) are promising tools for searching such large phase space. MI/ML models can be trained to screen candidate materials based on the location of their IR peaks, and atomistic simulations can be used to verify the validity of MI/ML predictions when the IR spectra of these potential candidates cannot be obtained experimentally yet. This underscores the significance of atomistic simulations in materials screening.
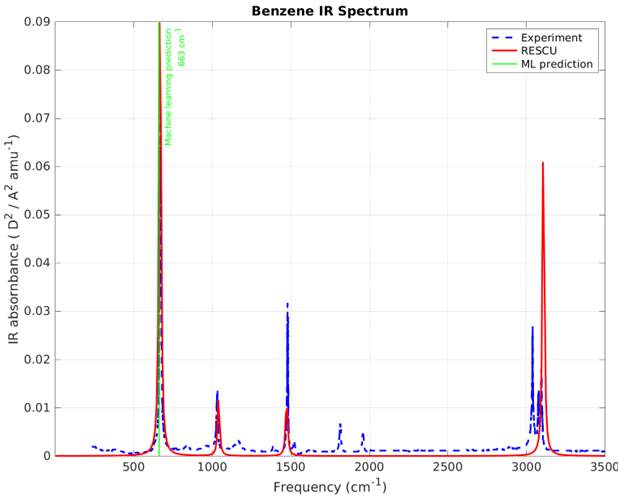
For example, we have built a machine learning model which can predict the location of the main IR peaks for input materials. As stated before, this information can be used to decide whether a given material is a good candidate for certain applications [4, 5], e.g., to be used in an IR detector for example. The model is trained based on the Random Forests method to ensure efficiency and accuracy, as validated by 5-fold cross-validation. The training data set is collected from public data resources which contains thousands or more of molecular and solid crystals with experimental/theoretical IR spectra. The atomic descriptors are generated based on geometrical atomic structures, chemical formulas as well as customized physical properties. RESCU is then used to determine the accuracy of the ML model by simulating the IR spectrum.
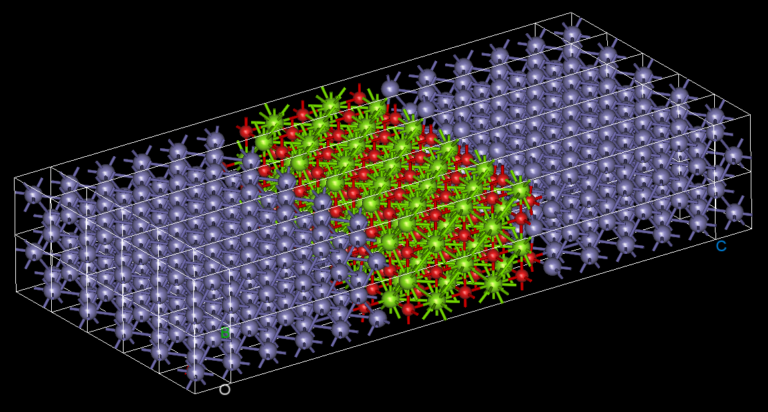
For newcomers to Nanoacademic’s atomistic solutions (Welcome!): What is RESCU? RESCU is a large-scale DFT code that provides an efficient and accurate IR spectrum simulator through its density functional perturbation theory (DFPT) module [1, 2, 3]. Please check out our previous posts on LinkedIn and on our website for relevant examples. The implementation is based on the 2n+1 theorem [6] and it can handle systems ranging from small molecules to solids, containing up to a few thousands of atoms. We have simulated the IR spectra of benzene molecule and beta-quartz crystals. Figures 2 and 4 show the IR spectra of these materials, which are in excellent agreement with the experiment. The ML predictions are also shown by light green lines in corresponding figures. The closeness of DFPT simulation and MI/ML predicted peaks guarantees the reliability of the MI/ML model for screening of materials based on their IR main peak location.

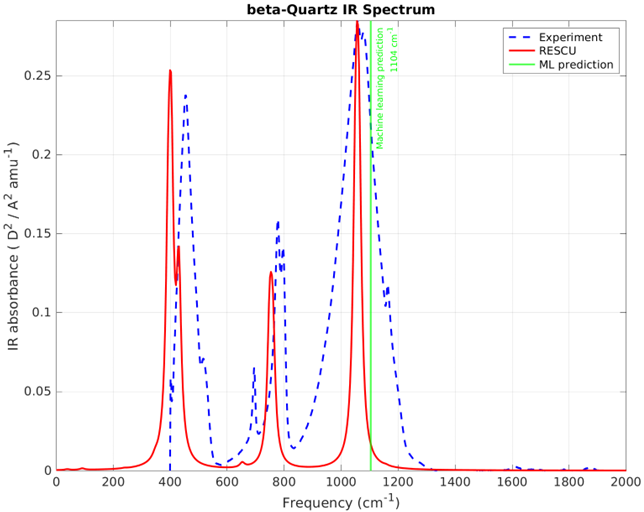
In this post, we showed how RESCU can be employed as an accurate tool for atomistic simulation of IR spectra for various types of materials, whether for a better understanding of available experimental data or as the sole solution to gain insight in cases where no such data is available. Indeed, both experimentalists and theorists find great benefits using our atomistic solutions.
Nanoacademic Technologies offers commercial RESCU licenses for single users and groups with different subscription possibilities and options. For more information, please check out our website and our documentation portal where you can also find more insights about RESCU’s features. For a 30-day free trial, please sign up on our user portal where you can download it. The ML method presented above is available for our community of researchers on a consultancy basis or through interfacing with their RESCU license to corroborate their experimental and simulated results which are obtained in their R&D projects. This is a truly innovative way to perform materials engineering by saving a lot of time and resources in the design process.
Please contact us for more information. See you soon!
Bibliography:
[1] Baroni, Stefano, Stefano De Gironcoli, Andrea Dal Corso, and Paolo Giannozzi. “Phonons and related crystal properties from density-functional perturbation theory.” Reviews of modern Physics 73, no. 2 (2001): 515. https://doi.org/10.1103/RevModPhys.73.515
[2] Xavier Gonze and Changyol Lee (1997). Dynamical matrices, Born effective charges, dielectric permittivity tensors, and interatomic force constants from density-functional perturbation theory: Phys. Rev. B 55, 10355 – Published 15 April 1997 https://doi.org/10.1103/PhysRevB.55.10355
[3] Bohloul, S. (2017). First-Principles Quantum Transport and Linear Response Modeling of Nano-devices and Materials. https://escholarship.mcgill.ca/concern/theses/8910jx167?locale=en
[4] Kang P, Liu Z, Abou-Rachid H, et al. Machine-learning assisted screening of energetic materials[J]. The Journal of Physical Chemistry A, 2020, 124(26): 5341-5351.
[5] Liu Z L, Kang P, Zhu Y, et al. Material informatics for layered high-TC superconductors[J]. APL Materials, 2020, 8(6): 061104.
[6] Nonlinear optical susceptibilities, Raman efficiencies, and electro-optic tensors from first-principles density functional perturbation theory, M. Veithen, X. Gonze, Ph. Ghosez, Phys. Rev. B 71, 125107 (2005) https://doi.org/10.1103/PhysRevB.71.125107
[7] Spectral Database for Organic Compounds SDBS https://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi
[8] RRUFF Project https://rruff.info/


