




By Dr. Mohammed Harb, Dr. Wanting Zhang, Dr. Vincent Michaud-Rioux and Mr. Jeremy F. Garaffa.
One of the most challenging steps in the process of simulating a material is adopting a description of its nucleus and electrons which is both:
i. Accurate
ii. Computationally efficient.
In this blog post available on LinkedIn as well, we will present a rapid overview of a few commonly used descriptions for nuclei, core electrons, and valence electrons and present our own newly optimized numerical atomic orbitals (NAO) for the Perdew-Burke-Ernzerhof (PBE) functional.
Nucleus and core electrons:
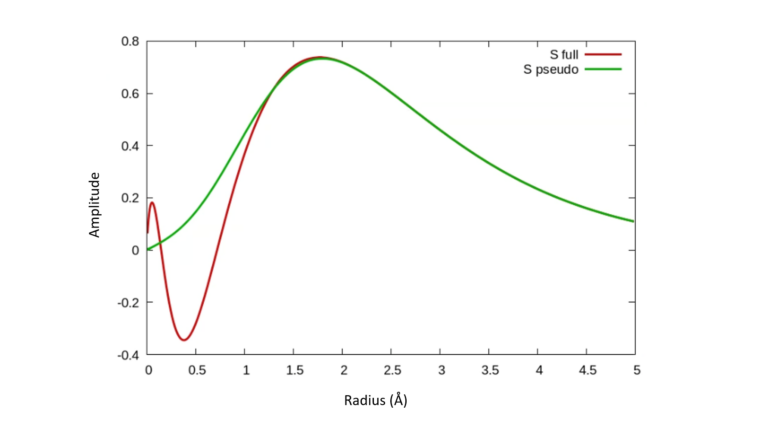
Core electrons are typically inert to the chemical environment due to their large binding energies. It is therefore possible to simplify their description by replacing the bare all-electron potential with a simpler “pseudopotential” – a term which captures the net effect of the core electrons and nucleus without the need for considering the singular Coulomb potential. The method also allows removing sharp features of valence wavefunctions near the origin. As an example, Figure 1 shows the pseudo-wavefunction of Si obtained from a pseudopotential and compares it to the wavefunction arising from the bare potential. The pseudo-wavefunction is better behaved and has less nodes and yet reproduces a charge density identical to the true wave function beyond a critical radius (~ 1 Å here).